Bi-specific Antibodies: The next-generation Antibody Therapeutics
Feb 13th 2020
Antibodies as therapeutic drugs has
significantly shifted the landscape of treatment in many areas of medicine
including oncology, viral infections and auto-immune diseases. Over the last two
decades, bispecific antibodies have created considerable interest as a new
approach in immunotherapy. As the name suggests, bispecific antibodies (bsAbs)
contain two different antigen-binding sites, capable of recognizing two
epitopes of an antigen or two separate antigens. These bispecifics can provide
a more robust immunogenic response via modulation of two different signaling
pathways in the same cell or co-engaging two different cells expressing either
antigens.
The concepts of bispecific antibodies were described as early as the 1960s, but it took more than two decades for bsAbs to be translated to the clinic. The first monoclonal bsAbs were generated in the 1980s by hybridoma technology, and the first article citing the therapeutic use of bsAbs was published in 1992. It has also been reported that bispecific IgG molecules are present in blood, placenta and milk (IgG and sIgA) in humans (Sedykh SE et al., 2012).
In 2009 catumaxomab (Trion Pharma), a rat-mouse hybrid bsAb for CD3 and EpCAM, was approved in Europe for the treatment of malignant ascites in people with EpCAM-positive cancer. Later in 2014, blinatumomab (Amgen) was approved for therapeutic use for refractory acute lymphoblastic leukemia by the US FDA. Currently, there are more than 100 bsAbs are under development, spanning a broad range of applications in diagnosis, imaging, prophylaxis, and therapy. However, most of the bispecifics are drug candidates for cancer therapy. Others are being developed for auto-immune diseases, Alzheimer’s disease, haemophilia, etc.
THE NEED FOR BSABS
Although
monoclonal Abs have been widely used as a therapeutic intervention in many
diseases especially cancer, they have certain limitations when it comes to
efficiency. One of the major challenge is low tumor penetration and retention
rate. It has been observed that in most patients only ~20% of the administered
dose binds to the target. Moreover, it has been observed that mAbs might block the
targeted pathways but somehow fail to induce sufficient immune responses
including T cell activation. ADCC CDC and ADCP are other mechanisms by which
mAbs can induce tumor cytotoxicity (Figure 1). In certain cases, relapse might
happen due to the extensive cross-talk among some signaling pathways in cancer
cells and nearby cells.

Figure 1: The schematic diagram of antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC) and antibody-dependent cell-mediated phagocytosis (ADCP) mechanisms in cancer treatment. ADCC involves natural killer (NK), whereas CDC is initiated when the C1q complex interacts with antibodies bound to the cell surface. ADCP involves recruitment of macrophages resulting in phagocytosis of the tumor cell. Reference: Wang et al., 2019
BsAbs present a range of opportunities to improve efficacy. They are usually designed to simultaneously bind to a tumor cell receptor and a cytotoxic immune cell, resulting in immune cell recruitment, commonly known as TRBAs (T-cell recruiting bi-specific antibodies). This functionality enables a positive cytotoxic immune cell activation and promotes better clearance of tumor cells. As an alternative to mAb combination therapy, bsAbs can also be designed to block two different receptors, increasing the anti-tumor effect, thus making it cost-efficient. Such dual immune-modulators more specifically exert their pharmacologic effects in the tumor area rather than a systemic immunomodulation. This considerably improves efficacy while minimizing systemic immune-related adverse effects.
A new class of bsAbs that have been conceptualized and are in the preclinical stages of development are the tumor-targeted immunomodulators. Unlike the TRBAs, these bsAbs selectively target and activate a smaller pool of tumor-specific T cells like the tumor infiltrating lymphocytes (TILs) by targeting a tumor antigen and costimulatory molecules such as CD40 or 4-1BB. These molecules have the capacity to induce a more robust T-cell cytotoxicity, better survival and long-term immunological memory (Dahlén et al., 2018).
EVOLUTION OF BSAB DESIGNS
Bispecific
antibodies were first described during the development of hybridoma technology.
Somatic fusion of two hybridomas gives rise to a ‘quadroma’ cell expressing two
different heavy and two different light chains that produce a variety of
different antibody species resulting from the random pairing of the heavy and
light chains. Catumaxomab (Removab, Trion Pharma), the first commercial bsAb
was produced by quadroma technology. It is a trifunctional Ab or a triomab with
HL fragments of mouse mAb against CD3 (IgG2a) and rat mAb (IgG2b) against
EpCAM, and the Fc which binds the Fc-receptor. The quadroma technology however needs
tedious purification strategies increasing its manufacturing costs.
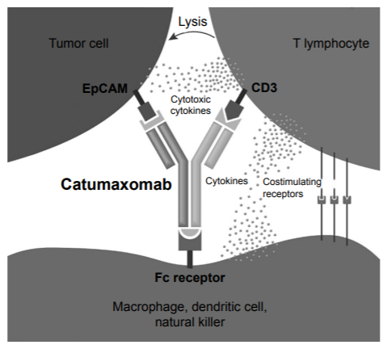
Figure 2: Mechanism of action of catumaxomab. Reference: Sedykh et al., 2018
Chemical conjugation was another conventional method in which two different purified mAbs are fused. This approach involved chemical cross linking of two different full-length IgGs or Fab fragments. Blinatumomab is one of the first generation bsAb drug designed in this format. Other approaches include formation of bispecific Fab antibodies using reduction and oxidation of sulfhydryl bonds processes and recombinant approaches to make bispecific antibody fragments based on single chain variable fragments (scFvs).
Over the last few years, the advent of genetic engineering has transformed bsAb research and development. A wide range of recombinant bsAb formats have been proposed, with over 50 different formats now available. It is now possible to create a ‘fit-for-purpose’ bsAb by adjusting the size, valency, flexibility, half-life and bio-distribution.
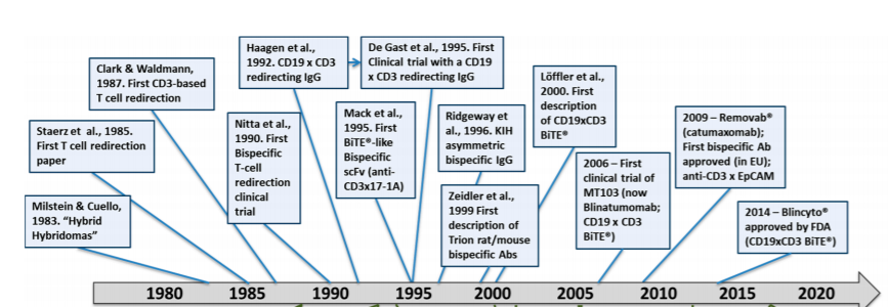
Figure 3: Key milestones in the history of T-cell redirected bispecific antibodies (TRBAs). Reference: Strohl and Naso, 2019
VARIOUS FORMATS OF BSABS AND THEIR MECHANISM OF ACTION
BsAbs or T-cell redirecting bsAbs (TRBAs) can be generally divided into two main categories: those with an Fc region (full length IgG based) or without an Fc region (the single chain variable fragment based, scFv). BsAbs with Fc region have a higher solubility and stability and can be purified easily using protocols established for IgG molecules. Another advantage of the presence of an Fc region is the induction Fc-mediated effector functions like ADCC, CDC and ADCP.
Antibody fragments, on the other hand, are smaller molecules with a short serum half-life. Unlike the extended pharmacokinetics of IgG molecules, these TRBAs require continuous dosing. Bispecific T-cell engager (BiTE), dual-affinity re-targeting proteins (DARTs) and Tandem diabodies (TandAbs) are some examples that come under this category. Conversely, the pharmacokinetic properties of scFv fragments can be improved by attachment of polyethyleneglycol groups or an Fc domain. Such ‘minibodies’ include Db-Fc fusion, taDb-Fc fusion or taDb-CH3 fusions. Additionally, there are some recently developed fragment-based bispecific antibody constructs, such as HSA-antibody fragment fusions, bispecific killer engagers (BiKEs), trispecific killer engagers (TriKEs) and dock-and-lock Fabs.

Figure 4: Architecture of common BsAbs formats. Reference: Strohl and Naso, 2019
(A) Bispecific T-cell engager (BiTE®)
(B) Dual affinity retargeting (DART®) antibody
(C) DART®-Fc for elongated half-life in vivo
(D) TCR fused to scFv called immune-mobilizing monoclonal TCRs against cancer (ImmTAC)
(E) mouse/rat hybrid IgG
(F) Asymmetric IgG with common LC (light chain)
(G) Asymmetric IgG with different light chains
(H) Asymmetric IgG-like molecule with a Fab arm and an scFv arm to eliminate light chain resorting
(I) Asymmetric IgG using crossmab technology for LC fidelity and extra Fab arm to make a 2:1 (target cell antigen:CD3ε) antibody call “TCBs,” for “T-cell Bispecifics”
(J) Chugai’s asymmetric IgG using “Asymmetric Reengineering Technology–Immunoglobulin” (ART-Ig®) platform technology, with an scFv fused to one HC to make an ART-Ig®-scFv 2:1 (target cell antigen:CDε) antibody
(K) Tetravalent, bispecific tandem diabody (TandAb)
(L) Tetravalent, bispecific ADAPTIR™ platform with two different scFvs fused to each Fc
MECHANISMS OF ACTION
The specificity and valency of the antigen binding sites on the bsAbs determine their mode of action. Bispecifics works by (Figure 5)
- a)Recruiting immune cells (T-cells or NK cells) to tumors. They will contain an antigen binding site specific to the tumor cell and to the immune cell. Examples are TrioMabs (catumaxomab), BiTEs (blinatumomab), DARTs and TandAbs.
- b)Blockade of signal receptors. Signaling pathways can be interfered by blocking two or more receptors simultaneously. Examples include DVD-Igs, DAFs, 2-in1-IgG, Tv-IgGs and CrossMabs.
- c)A recent and exciting application is BsAb mediated forced assembly of the coagulation Xase complex. A heterodimeric common light chain IgG connects FXIa and FX (Factor X) and there by overcomes FVIII deficiency.

Figure 5: Different modes of action of therapeutic bispecific antibodies. Abbreviations: BiTE, bispecific T cell engager; DAF, dual-action Fab; DART, Dual affinity retargeting; DNL, dock-and-lock; DVD-Ig, dual variable domain immunoglobulins; FX, Factor X; HSA, human serum albumin; kih, knobs-into-holes; Tv, tetravalent. Reference: Kontermann and Brinkmann, 2015
BISPECIFIC T-CELL ENGAGERS (BITES)
BiTE molecules have been successfully used as immunotherapy for redirecting T cells towards tumor cells. The scFv fragments from two different monoclonal antibodies are connected by a short flexible peptide linker that enables free rotation of the two arms. This flexibility is essential for targeting the receptors on the cell membranes of the cytotoxic T-cell and tumor cell; and the subsequent activation of the T cell.
Blinatumomab (Blincyto, Amgen) was the first BiTE drug that was approved by the FDA for the treatment of B-cell precursor acute lymphoblastic leukemia (ALL) (Figure 6). The small size of the molecule helps it to attain a close proximity to T-cell and target cell membranes, but on the downside this feature leads to the rapid clearance from circulation. For this reason BiTE requires continuous dosing at a high concentration (15–28 µg per day) to recruit and activate a large amount of suboptimal T cells to achieve half-maximal target cell lysis (Wang et al., 2019).
More than 15 BiTEs are currently being tested in clinical trials, few of which are the next-generation half-life extended BiTEs i.e. BiTE-Fcs.
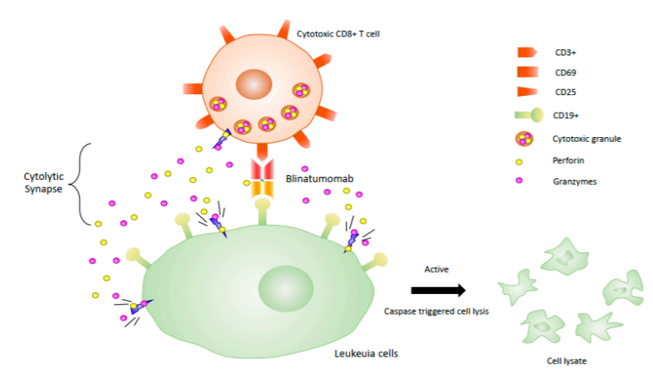
Figure 6: Different modes of action of therapeutic bispecific antibodies. Abbreviations: BiTE, bispecific T cell engager; DAF, dual-action Fab; DART, Dual affinity retargeting; DNL, dock-and-lock; DVD-Ig, dual variable domain immunoglobulins; FX, Factor X; HSA, human serum albumin; kih, knobs-into-holes; Tv, tetravalent. Reference: Kontermann and Brinkmann, 2015
DUAL-AFFINITY RE-TARGETING PROTEINS (DARTS)
DARTs were primarily designed to overcome the shortcomings of BiTEs. DART comprises of two Fv fragments, with two unique antigen-binding sites formed when two Fv fragments heterodimerize. The Fv1 consists of a VH from antibody A and a VL from antibody B, while Fv2 is made from a VH from antibody B and VL from antibody A i.e. in a VL (1)-VH(2) and VL (2)-VH(1) order (Figure 7). This combination allows DART to mimic natural interaction within an IgG molecule. Short linker sequences between the VL and VH segments promote a posttranslational “diabody”-type association. The interchain linkers and the covalent linkage between the two DART chains limits the freedom of the antigen binding domains, resulting in a stable association between target and effector cells.

Figure 7: DART molecule. Reference: Wang et al., 2019
DART molecules can be produced easily in mammalian expression systems without the need for a refolding process. When compared to BiTEs, DART molecules have shown to be more stable and potent. In an experiment by Moore et al. to compare the in vitro ability of CD19xCD3 DART and BiTE molecules, DART molecules were found to be more consistent than BiTE molecules in targeting and killing B-cell lymphoma.
Several DART molecules are under development for T-cell redirection, modulation of receptor signaling and neutralization of viruses (Figure 8).

Figure 8: Different DART molecules in preclinical and clinical development. DART molecules can have a variety of functions, including redirection of T-cell–mediated cytolysis (MGD006/S80880, MGD007, MGD011, and MGD014), modulation of receptor signaling (MGD010), and neutralization of viruses.
Reference: https://www.medicographia.com/2018/01/the-versati...
TANDEM DIABODIES (TANDABS)
TandAbs are tetravalent with four binding sites, two for tumor antigens and the other two to bind to immune cells. TandAbs do not have an Fc domains and are smaller than IgGs, hence have a shorter serum half-life than IgGs but larger than BiTEs.
AFM13 and AFM11 (developed by Affimed) are two TandAbs currently in clinical trials for patients with Hodgkin’s disease and non-Hodgkin’s lymphoma respectively. AFM13 is an NK cell recruiter that binds to CD30 and CD16A on NKcells and macrophages. AFM11, in contrast, is a T-cell recruiter that binds to CD3 on T-cells and CD19 on lymphomas, a mode of action similar to blinatumomab.
EXAMPLES OF OTHER BSABS FORMATS UNDER DEVELOPMENT
| Sl.
No |
BsAb Format | Structure | Targets | Mode of Action | Manufacturer |
| 1 | (scFv)2-HAS and Tetravalent-IgG derivatives(Tv-IgGs) |  |
Her2 + Her3 or IGF1R + Her3 | Blocks intracellular signaling cascades involving HER family proteins to suppress tumor growth. | Merrimack |
| 2 | Dual-action Fab (DAF)-IgG |  |
HER1+HER3 | Complete inhibition of mitogen- activated protein kinase (MAPK) and AKT signaling to arrest tumor growth. | Genentech |
| 3 | H-chain heterodimers with orthogonal Fab interfaces |  |
HER1 and c-Met | Dual blockade of HER1 and c-MET that have a role in tumor growth and metastasis. | EliLilly |
| 4 | DVD-IgG |  |
TNF + IL-17 and
IL1a + IL1b |
Simultaneous inhibition of inflammatory signal pathways. Being tested for rheumatoid arthritis (RA). | Abbott and Abbvie |
| 5 | Bi-nanobodies |  |
IL-17A+IL-17F | Blocks inflammatory signal pathways to treat autoimmune diseases. | Merck Serono – Ablynx |
| 6 | Crossmabs |  |
VEGFA and Angiopoetin 2 | Targets 2 or more angiogenic factors simultaneously to improve anti-tumor effect. | Roche |
| 7 | TBTI(DVD)-IgG | Tetravalent bispecific tandem Ig similar in composition to DVD-Igs | IL4 and IL13 | Reduce IL4 and IL13 dependent fibroblast activation. Suitable for treating fibrotic diseases like IPF | Sanofi |
| 8 | ScFv-IgGs | 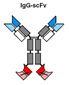 |
L17 and IL23 | Block L17 and IL23 signaling to treat inflammation and auto-immune diseases. | BMS–Zymogenics |
KNOB INTO HOLES TECHNOLOGY
First proposed by Francis Crick and later applied by Ridgeway et al. in Fc engineering, the knobs-into-holes technology was a breakthrough in the generation of bivalent, bispecific IgG molecules. Here, the yield of heterodimers from two different heavy chains is increased introducing different mutations into the two CH3 domains. One heavy chain will have the T366W mutation (“knob”: replacement with a sterically bulky amino acid) which joins with the second heavy chain with mutations T366S, L368A, or Y407V (“hole”: replacement by a smaller amino acid); forming a heterodimer that is thermodynamically stable.

Figure 9: KIH scaffolds. Two different scFv (in circles) are anchored on Fc (K) and Fc (H), respectively, defined as scFvKIH or scFv-KIHr (the reversed arrangement of scFv-KIH). Alternatively, 2 scFv are connected with a flexible linker, resulting in a BiTE. BiTE-KIH is defined as BiTE on Fc (K) and BiTE-KIHr is as BiTE on Fc (H). The pairing Fc (H) or Fc (K) contains the hinge, CH2 and CH3 domains only. Examples are shown for diL2K (anti-CD3) and 5–10 (anti-EpCAM).
ADVERSE EFFECTS
Similar to other monoclonal antibody treatments, therapeutic BsAbs cause various side effects like nausea, vomiting, abdominal pain, fatigue, leukopenia, neutropenia, and thrombopenia. In many patients, Abs against therapeutic BsAbs appear in the blood during treatment. Adverse events often occur during the start of the therapy and tend to normalize on continued treatment.
A common side effect seen with blinatumomab and catumaxomab therapy is “cytokine storm”, an elevation of cytokine levels due to the engagement of cytotoxic T-cells. A low initial dosing and subsequent increase in doses along with corticosteroids and anti-histamines can help lessen these side effects. Hepatoxicity is also a common adverse event seen in patients undergoing blinatumomab treatment.
Other notable adverse events that have been reported include neurological and psychiatric side effects which are usually completely reversible after the end of therapy; and bone marrow toxicity.
THE WAY FORWARD
TRBAs have been quite successful in the treatment of hematological malignancies and show significant promise for use in many forms of cancer. Simultaneous blocking of several biological pathways using BsAbs have demonstrated synergistic therapeutic effects unachievable with a mixture of monospecific Abs.
One of the main challenges in this area is the manufacturing of bsAbs. There are certain challenges to produce uniform bispecific antibody with high quality and limited or negligible side products and impurities. Selecting the appropriate host expression system depends on the specific scFv antibody size, amino acid sequence, protein conformation, solubility, stability, purification, and scalability. The protein stability and tissue penetration ability vary and depend on different types of scFv antibody. Purification of IgG is much easier compared to Ab-fragments. For IgG-like full-size bispecific antibody, the production of pure heterodimer is achieved by complete heavy chain and light-chain heterodimerizations.
With numerous bsAbs entering clinical trials, there is a need for thorough analysis of the pharmacokinetic properties and mechanisms of action. The ideal therapeutic BsAbs are expected to have a long half-life in human blood, distribution among organs, and sufficient penetration of tissue. Another point to be considered is that the affinity between the T-cell antigen arms should be lower than the affinity of the second arm to the tumor antigen. This is because higher affinity on the immune cell antigen arm can cause higher distribution of the bsAbs in T-cell rich tissues such as lymph node and spleen rather than into tumor tissues and might lead to toxicity.
The last two decades has seen a surge in immunotherapy research with several immunotherapeutic products being approved for various diseases. Immunomodulation using monoclonal antibodies, bispecific antibodies and CAR T-cells have shown great promise and is revolutionising cancer treatment. All these modes of therapy possess their own advantages and challenges. Currently, cost of treatment favours TRBAs whereas T cell therapy shows a better duration of response. However, technological advancements in phage display screening, antibody linker engineering, quadroma technology, knobs-into-holes technology, common light chain strategy, CrossMAb technology and protein engineering have boosted the development of bispecific antibodies and their derivatives. Application of bispecific antibodies have extended other diseases such as viral infections, AIDS, genetic diseases and also diagnostics. Bispecific antibodies is undoubtedly a promising therapeutic strategy; and will definitely be a complementary approach to the conventional therapies in the near future.
REFERENCES
Eva Dahlén, Niina Veitonmäki and Per Norlén. Bispecific antibodies in cancer immunotherapy. Therapeutic Advances in Vaccines and Immunotherapy 2018, Vol. 6(1) 3–17 DOI: 10.1177/ 2515135518763280.
G. R. Chichili, P. A. Moore, S. Johnson and E. Bonvini. The versatility of the DART® platform for multiple therapeutic applications. www.medicographia.com
Nicolas Fischer, Olivier Léger. Bispecific Antibodies: Molecules That Enable Novel Therapeutic Strategies. Pathobiology 2007; 74:3–14 DOI: 10.1159/000101046.
Qiong Wang, Yiqun Chen , Jaeyoung Park , Xiao Liu, Yifeng Hu, Tiexin Wang, Kevin McFarland and Michael J. Betenbaugh. Design and Production of Bispecific Antibodies. Antibodies 2019, 8, 43; doi:10.3390/antib8030043.
Roland E. Kontermann, Ulrich Brinkmann. Bispecific antibodies. Drug Discovery Today 20 (July (7)) (2015) 838–847.
Roland E. Kontermann, Ulrich Brinkmann. Corrigendum to “Bispecific antibodies” [Drug Discov. Today 20 (July (7)) (2015) 838–847]. Drug Discovery Today, Volume 24, Issue 7, July 2019, Pages 1422
Sedykh SE, Buneva VN, Nevinsky GA. Human milk sIgA molecules contain various combinations of different antigen-binding sites resulting in a multiple binding specificity of antibodies and enzymatic activities of abzymes. PLoS One. 2012;7(11):e48756. doi:10.1371/journal. pone.0048756
Sergey E Sedykh, Victor V Prinz, Valentina N Buneva, Georgy A Nevinsky. Bispecific antibodies: design, therapy, perspectives. Drug Design, Development and Therapy 2018:12 195–208.
William R. Strohl and Michael Naso. Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells. Antibodies 2019, 8, 41; doi:10.3390/antib8030041.
Yiren Xu, John Lee, Cuong Tran, Tyler H Heibeck, Willie D Wang, Junhao Yang, Ryan L Stafford, Alexander R Steiner, Aaron K Sato, Trevor J Hallam, and Gang Yin. Production of bispecific antibodies in “knobsinto-holes” using a cell-free expression system. mAbs 7:1, 231--242; January/February 2015.
Written By: Shalitha Sasi