How Far We Have Advanced in the Chemotherapy World
Feb 26th 2020
Cancer treatment is the single most challenging endeavor for scientists in the biomedical field. With the current non-selective chemotherapy approaches, the patient is at a high risk of serious post treatment complications. Conventional chemotherapy methods kill a large population of cells in a normal body with the hope to have the cancer cells terminated quicker than normal biologics. Unfortunately, other than radiotherapy, more often such non-selective treatments are the only hope for the suffering patient.
This leaves a gap and an urgent need for development of a novel method that could specifically target cancer cells and leave the normal ones unharmed. Inspired by an early work in the biotechnology, monoclonal antibodies attracted scientists to serve as a Trojan horse to selectively pass through the gates of cancer cell walls, and smuggle in cytotoxins that are necessary to terminate affected lesions. Cell surface is a very rich and dynamic environment coated with receptors that act as communication satellites. They are used in many vital cellular functions such as cell-cell recognition, signal transduction, extracellular matrix secretion and other survival mechanisms. Cell membranes are highly protective and discriminatory to ensure cell integrity remains intact. Fortunately, cancer cells tend to display unique receptors on their surface, which could be easily recognized by specific antibodies in a heterogeneous environment. Such communications allow for an unrestrained VIP access into cytoplasm through receptor-antibody interactions. Once the antibody passes through the checkpoints of cell membrane, it could release a toxin to kill the deceived cancer cell. This technique has since advanced significantly to arm physicians with an extraordinary weapon to tackle cancer growth rate.
Nowadays, scientists are able to isolate a particular cancer cell receptor (antigen) and engineer a monoclonal antibody to which a highly potent cytotoxic agents are attached using chemical linkers. This targeted drug delivery system effectively couples the precision of the antibody targeting moiety with the cytocidal activity of the payload, which is generally too toxic on its own to be systemically administered to patients. Such approach reduces the off-target toxicities in patients by restricting the exposure of normal tissues to the payload, hence broadening the potential therapeutic window compared with traditional chemotherapy. Immunotherapy has shown such promising results that the number of investigational agents in human trials has tripled over the past 5 years alone. Antibody-Drug Conjugate field continues to evolve to improve on target selectivity, multivariable cytotoxin payloads and bio-conjugation methods.

Herein we introduce the three different aspects of engineering an Antibody-Drug Conjugate (ADC) molecule. Despite a plethora of tumor associated antigens that have been identified on malignant cells for immunotherapy based applications, only a small suit of binding sites are useful for sustainable therapy. Ideally, the target antigen should be expressed abundantly on cancerous cell surface and most importantly, demonstrate substantial differential distribution pattern relative to normal tissues to reduce off-target toxicities. Unfortunately, current identified antigens only marginally meet the criteria, and most ADCs currently in development are at best preferentially expressed in malignant cells, exhibit low expression on normal cells, and/or are absent in vital or regenerative tissues. Contrary to this criterion, another challenge the scientific community faces is to engineer a complex that maintains a certain binding affinity not too high to prevent toxicity of ADC over normal cells.
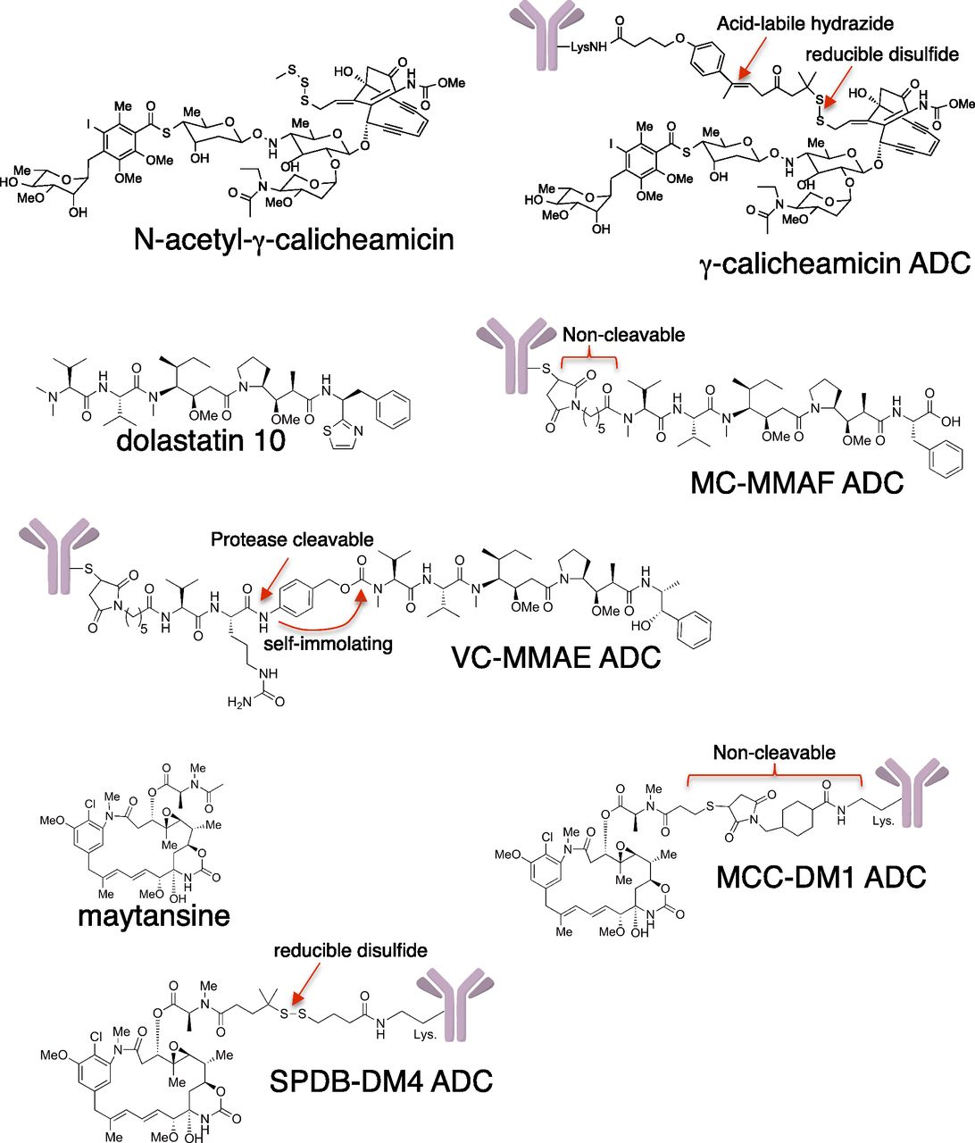
Second challenge is to design a complex that allows for the internalization of ADC into the cellular environment to enable intracellular delivery of the payload. Thus, the endocytic properties of the target are a key determinant in the selection of an appropriate antigen. There are multiple mechanisms through which an ADC could release its payload once successfully entered into cell. Since there is a covalent linkage between the drug and the antibody, a cleavable linkage is often used to facilitate the release. The linker should be stable enough to survive the circulation and allow the cytotoxic moiety to remain attached to the antibody as it is distributed into tumor tissues, yet permit efficient release once internalization and trafficking in specific subcellular compartments has occurred. Linkers with reduced stability are prone to non-specific cleavage, which contributes to higher systemic levels of drug and a broader toxicity profile. Cleavable linkers are usually susceptible to hydrolysis, pH ranges and enzymatic cleavage inside the cell. Non-cleavable linkers are more robust and do not simply break upon delivery so they need harsher cellular intervention through the aid of lysosomal proteolytic degradation of the antibody and release the payload as an active metabolite.
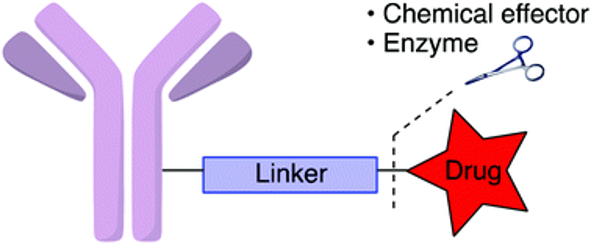
Finally, the effector component of any ADC is the drug itself and should satisfy certain requirements including but not limited to potency at sub-nanomolar ranges, structural feasibility for conjugation and solubility/stability to deliver the purpose in different cellular environment. The very first FDA approved ADC was Doxorubicin that despite initial promises, displayed insufficient potency in human subjects so was recognized as clinically ineffective. Despite such inadequacies, there are still many payloads that are suitable and appropriate for conjugation to an antibody complex. An additional challenge is to account for the tumor cell’s proteolytic enzymes or even extracellular catalytic biomolecules that could cause premature release of the cytotoxin to undesired areas. Examples of payloads are Tubulin inhibitors such as auristatins and maytansinoids or DNA damaging agents.
Auristatins are synthetic analogs of the natural product dolastatin-10, originally isolated from the sea hare Dolabella auricularia. It interferes with cell cycle by inhibiting the tubulin polymerization to cause cellular apoptosis. Auristatins is 20-50 times more potent than that of other family members of tubulin-interacting agents, such as vinblastine. Other payloads in the form of peptide analogs of dolastatin-10 Monomethyl Auristatin-E (MMAE) and Monomethyl Auristatin-F (MMAF), have also demonstrated great efficacy in the ADC world. Since MMAE is membrane permeable, it could easily diffuse from antigen-positive tumor cells into neighboring cells and act as a by-standard killer to terminate affected cell in an antigen-independent manner. In contrast, MMAF suffers from higher hydrophilicity hence cannot pass though the cell membrane as readily, leaving the ADC complexes less effective in vivo than those with MMAE. The FDA approved CD30-targeting ADC Brentuximab Vedotin received an accelerated approval to Biologics License Application (BLA) for the treatment of relapsed Hodgkin lymphoma (HL) and systemic anaplastic large cell lymphoma (ALCA). Later in 2017, the approval was extended for the treatment of cutaneous T-cell lymphoma (CTCL) patients, who have received prior systemic therapy. Another class of antimitotic tubulin inhibitors are Maytansinoids that are derived from maytansine, a natural product isolated from the bark of African shrubs. Unusual high potency of maytansine made them the first compound to be discovered in the picomolar IC50 values and potency up to 1000-fold more than that of tubulin inhibitors such as Paclitaxel. Through a semi-synthesis manipulation, a series of maytansine analogs were developed that were suitable for covalent linkage with antibodies including. Example of such ADCs are payload used in the approved agent ado-trastuzumab mertansine. Another big class of payloads are derived from calicheamicin which is a potent antibiotic that binds the minor groove of DNA to induce DNA double-strand cleavage and cause cell death. The initial clinical results of ADCs coupled to DNA damaging agents were unsatisfactory due to small therapeutic indices and serious late toxic effects, but most of these concerns have been addressed in the newer generation of this class by improving on the chemical bonding to antibody. Two of the four currently approved ADCs (gemtuzumab ozogamicin and inotuzumab ozogamicin) carry a payload from this family and are commonly used in clinical treatments. That is derivative of calicheamicin, a potent antibiotic that binds the minor groove of DNA to induce DNA double-strand cleavage.
There are currently more than 80 ADC related compounds in various stages of clinical development, that shape the fast growing field of cancer therapy. ADCs continue to be the single most promising approach with significant investment and the ambition to selectively deliver cytotoxic agents to cancer cells with minimum toxicity to normal cell population. Although clinical gaps still exist in the optimal application of ADCs in oncology, the study of these agents in a variety of settings is harnessing novel technologies and leveraging translational medicine to maximize the therapeutic index of these agents. Clinical development strategies will include alternative dosing schedules and cutting-edge translational medicine to optimize patient selection, capture response signals early, match biomarkers to warhead mechanisms of action, and evaluate potential combination therapies to maximize the therapeutic index of ADCs. By incorporating these novel technologies and biomarker selection strategies, ADCs will be well positioned to provide clinical benefit to a much broader patient population and contribute to the future of therapeutics of what we will eventually call personalized medicine.
Written by ChemVice Inc.
References:
- Moek KL, de Groot DJA, de Vries EGE, et al. The antibody-drug conjugate target landscape across a broad range of tumour types. Ann Oncol 2017;28(12):3083-3091.
- Chari RV, Miller ML, Widdison WC. Antibody-drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed Engl 2014;53(15):3796-827.
- Trail PA, Dubowchik GM, Lowinger TB. Antibody drug conjugates for treatment of breast cancer: Novel targets and diverse approaches in ADC design. PharmacolTher 2018;181:126-142
- Szot C, Saha S, Zhang XM, et al. Tumor stroma-targeted antibody-drug conjugate triggers localized anticancer drug release. J ClinInvest 2018;128(7):2927- 2943
- Donaghy H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs 2016;8(4):659-71.
- Tsuchikama K, An Z. Antibody-drug conjugates: recent advances in conjugation and linker chemistries. Protein Cell 2018;9(1):33-46.
- McCombs JR, Owen SC. Antibody drug conjugates: design and selection of linker, payload and conjugation chemistry. AAPS J 2015;17(2):339-51.
- Chen H, Lin Z, Arnst KE, et al. Tubulin Inhibitor-Based Antibody-Drug Conjugates for Cancer Therapy. Molecules 2017;22(8)
- Chari RV. Expanding the Reach of Antibody-Drug Conjugates. ACS Med ChemLett 2016;7(11):974-976
- Beck A, Goetsch L, Dumontet C, et al. Strategies and challenges for the next generation of antibody-drug conjugates. NatRevDrugDiscov 2017;16(5):315-337.